News
Apr 22, 2026
Ehsaneh’s paper on cholesterol-mediated membrane modulation has been recognized as a Highly Accessed Paper. Congratulations!
Apr 10, 2026
Nicholas Henderson co-authored a paper published in Nature Communications. Congratulations!
Apr 8, 2026
Congratulation Asal Behzadnezhad on passing the candidacy exam!
Apr 3, 2026
Professor Moradi’s research on physics-informed AI has been recognized by the National AI Research Resource (NAIRR) and featured in its Science Highlights, highlighting the impact of his work.
Mar 21, 2026
Matthew successfully defended his Ph.D. Congratulations, Dr. Matthew Brownd!
Feb 23, 2026
Ehsaneh, Soheil, and Saeid gave a talk at the BPS 2026 meeting in San Francisco, CA.
Feb 21, 2026
Moradi Lab attended Biophysical Society annual meeting in San Francisco, CA.
Jan 7, 2026
Stephanie Sauve received the “The George D. Blyholder Endowed Award in Physical Chemistry” for the 2025-2026 school year. Congratulations Stephanie!
Jan 6, 2026
Stephanie Sauve's paper was published in The Journal of Physical Chemistry B. Congratulations Stephanie!
Aug 29, 2025
Ehsan Khodadadi's paper was published in The Membranes. Congratulations Ehsan!
Jun 25, 2025
Ehsaneh Khodadadi's paper was published in The Membranes. Congratulations Ehsaneh!
Apr 24, 2025
Ehsaneh successfully defended her Ph.D. Congratulations, Dr. Ehsaneh Khodadadi!
Mar 24, 2025
Ehsan gave a talk at the 2025 ACS Conference in San Diego, CA, titled 'Computational Investigation of Cholesterol Dynamics in Liposomal Drug Delivery Systems: A Coarse-Grained Simulation Study.'
Mar 23, 2025
Ehsaneh gave a talk at the 2025 ACS Conference in San Diego, CA, titled 'Influence of Cholesterol on the Activation Mechanism of mGluR2,' focusing on protein-lipid interactions in different functional states.
Feb 15, 2025
Ehsaneh presented a flash talk at BPS 2025 in Los Angeles, CA.
Feb 15, 2025
Ehsan Khodadadi presented a flash talk at BPS 2025 in Los Angeles.
Feb 15, 2025
Moradi Lab attended Biophysical Society annual meeting in Los Angeles, CA.
Dec 16, 2024
Stephanie Sauve and Hope Woods Shaw attended the 62nd Hands-On Workshop on Computational Biophysics at Auburn University in Auburn, AL.
Nov 12, 2024
congratulation Stephanie Sauve on passing the candidacy exam and being a PhD candidate in the Cell and Molecular Biology program at the University of Arkansas
Nov 05, 2024
Shadi and Vivek's paper titled "Molecular Dynamics Investigation of the Influenza Hemagglutinin Conformational Changes in Acidic pH" was published in the Journal of Physical Chemistry B. Congratulations to Shadi, Vivek, and Prof. Moradi!
Nov 01, 2024
Prof. Mahmoud Moradi Interviewed With World Chemistry, the Royal Society of Chemistry's official news magazine:Dr. Mahmoud Moradi shared insights on protein simulation in physiological environments, spotlighting advances in structural biology and drug design.
Sep 20, 2024
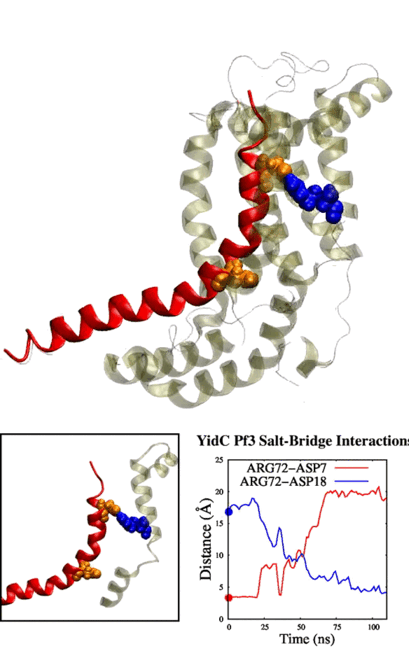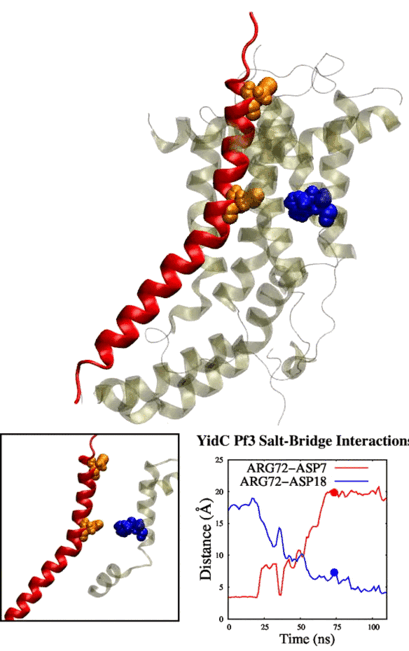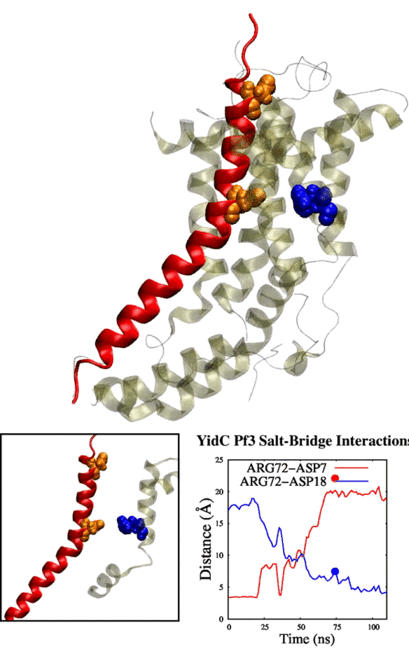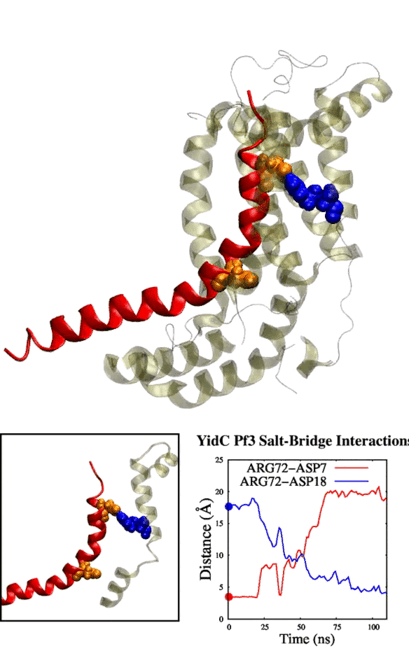
Adithya and Shadi's paper titled "Deciphering the Inter-domain Coupling in a Gram-negative Bacterial Membrane Insertase" was accepted in The Journal of Physical Chemistry B. Congratulations Adithya and Shadi!
Aug 22, 2024
Ugochi and Adithya's paper titled "Differential Behavior of Conformational Dynamics in Active and Inactive States of Cannabinoid Receptor 1" was published in The Journal of Physical Chemistry B. Congratulations Ugochi and Adithya!
Aug 15, 2024
Saeid Ghomi joined the lab as a Ph.D. student in the Computer Engineering, Welcome, Saeid!
Jul 18, 2024
Shadi successfully defended her Ph.D. Congratulations, Dr.Shadi Badiee!
Jul 9, 2024
July 1, 2024, Mahmoud Moradi appointed as the Bruker Analytical Science Professor of Chemistry and Biochemistry.
Jun 6, 2024
Ehsaneh presented at the Molecular Biophysics of Membranes conference held in Tahoe, California, USA, from June 2-7, 2024.
May 15, 2024
Nicholas Henderson joined the lab as a Ph.D. student in the Chemistry. Welcome, Nicholas!
May 15, 2024
Milad Sangsefidi joined the lab as a Ph.D. student in the Mechanical Engineering, Welcome, Milad!
Apr 30, 2024
Ugochi successfully defended her Ph.D. Congratulations, Dr. Ugochi Isu!
Apr 16, 2024
Professor Moradi wins the Fulbright College Outstanding Researcher Award for the 2023/24 academic year.
Apr 15, 2024
Ugochi Isu was selected as a D.E Shaw Research (DESRES) Doctoral & Postdoctoral Fellow. She attended a symposium in New York City on April 4-5th, sponsored entirely by DESRES.
Feb 19, 2024
Ugochi Isu gave a talk at the Biophysical Society's annual meeting in Philadelphia, PA.
Feb 19, 2024
Soheil gave a talk at the Biophysical Society's annual meeting in Philadelphia, PA.
Feb 16, 2024
Moradi Lab attended Biophysical Society annual meeting in Philadelphia, PA.
Jan 16, 2024
A paper Stephanie contributed called "Bioorthogonal click labeling of an amber-free HIV-1 provirus for in-virus single molecule imaging" was Published!
Cell Chemical Biology Congrats!
Asal Behzadnezhad joined the lab as a Ph.D student in the CEMB. Welcome, Asal!
Jan 4, 2024
Reza Taghavi joined the lab as a Ph.D student in the MSEN. Welcome, Reza!
Jan 3, 2024
Dr.Hope Woods joins the lab as a postdoctoral fellow. Welcome Hope!
Dec 1, 2023
Ehsaneh Won First Place in MSEN Poster Presentation Contest. Congrats Ehsaneh!
Jun 29, 2023
Goolsby, Curtis; Losey, James; Fakharzadeh, Ashkan; Xu, Yuchen; Düker, Marie-Christine; Getmansky Sherman, Mila; Matteson, David; Moradi, Mahmoud's paper called ” Addressing the Embeddability Problem in Transition Rate Estimation " was Published!
The Journal of Physical Chemistry Congrats all!
M Benton, M Furr, V Govind Kumar, A Polasa, F Gao, C-D Heyes, T-K Suresh Kumar, M Moradi's paper called ” cpSRP43 is both highly flexible and stable: Structural insights using a combined experimental and computational approach " was published!
Journal of Chemical Information and Modeling Congrats all!
Shadi, Ugochi, and Ehsaneh’s paper called "The Alternating Access Mechanism in Mammalian Multidrug Resistance Transporters and Their Bacterial Homologs" was punblished in Membranes!
Membranes Co$
Stephanie, Joseph, and Adithya's paper on the rocker-switch mechanism in the major facilitator superfamily was selected as the cover of Membranes!
Membranes Congrats all!
Ahmed Shubbar joined the lab. Welcome Ahmed!
May 8, 2023
Ehsan Khodadadi joined the lab. Welcome Ehsan!
May 3, 2023
Ugochi defended her masters in Cellular and Molecular Biology. Congrats Ugochi!
May 1, 2023
Samuel Wamwere Mwatha joined the lab. Welcome Samuel!
Apr 25, 2023
Stephanie, Joseph, and Adithya's paper called "Ins and Outs of Rocker Switch Mechanism in Major Facilitator Superfamily of Transporters" was punblished in Membranes!
Membranes Co$
Ugochi, Shadi, and Ehsaneh's paper called "Cholesterol in Class C GPCRs:Role, Relevance, and Localization" was punblished in Membranes!
Membranes Congrats Ugochi, Shadi, and Ehsaneh!
Group dinner at Pacific Catch in La Jolla, CA.
Feb 18-23, 2023
The lab attended the 2023 Biophysical Society Annual Meeting in San Diego, CA!
Jan 16, 2023
Adithya's paper called "Towards A Purely Physics-based Computational Binding Affinity Estimation"" was published in Nature Computational Science.
Nature Computational Science Congrats Adithya!