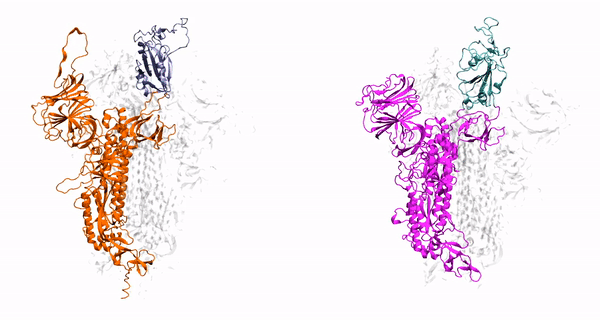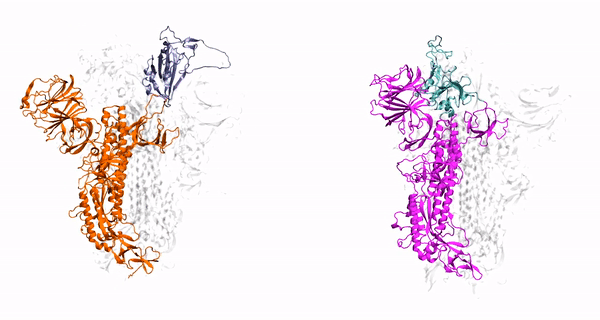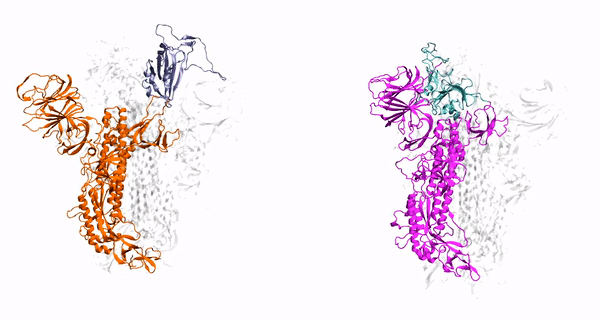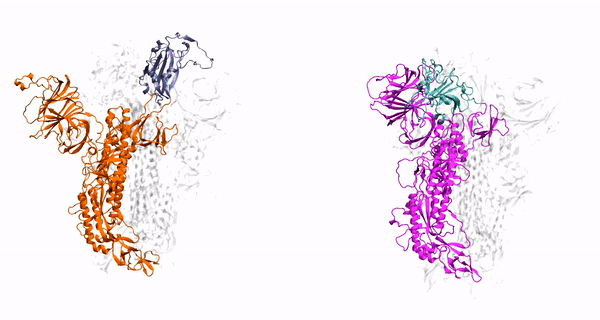
Cholesterol Dependence on the Conformational Changes of Metabotropic Glutamate Receptor 1 (mGluR1)
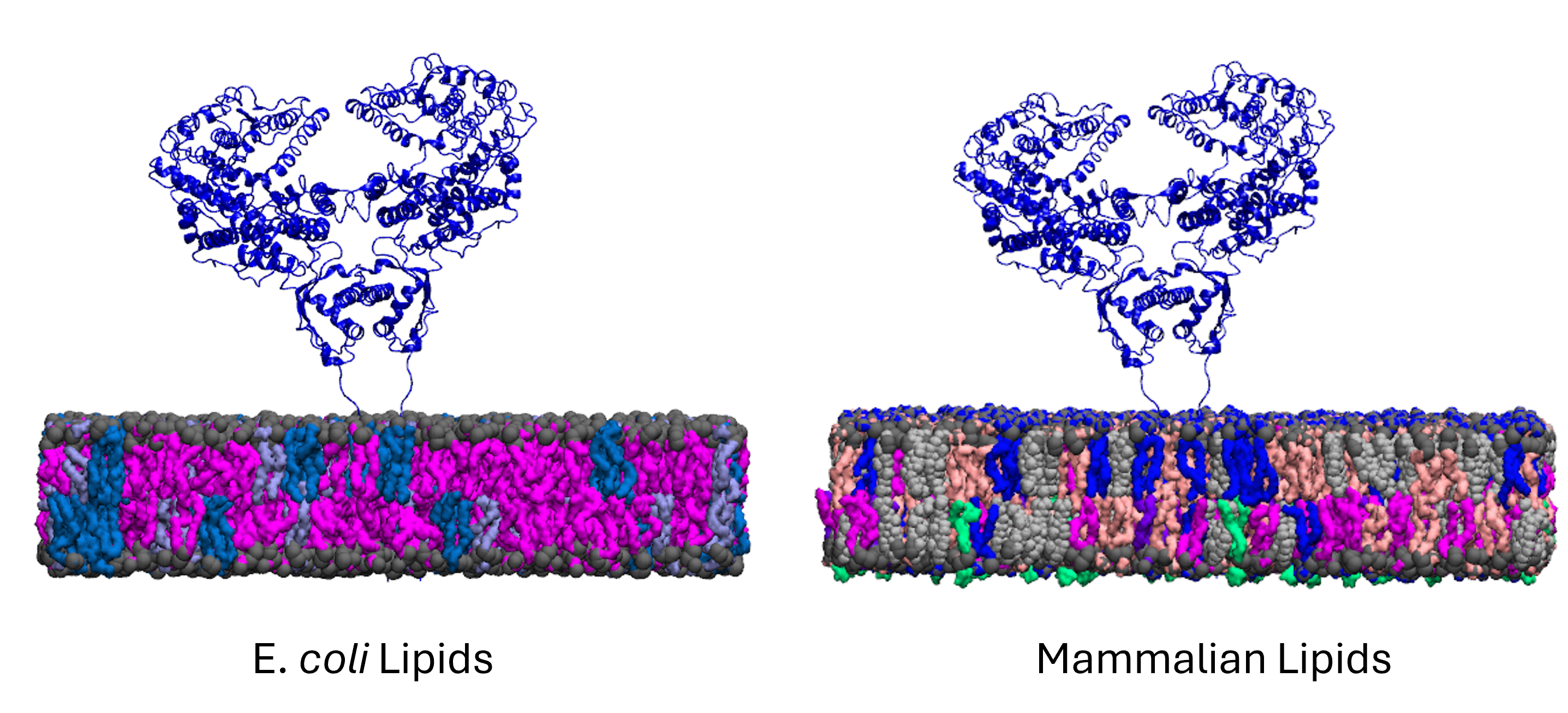
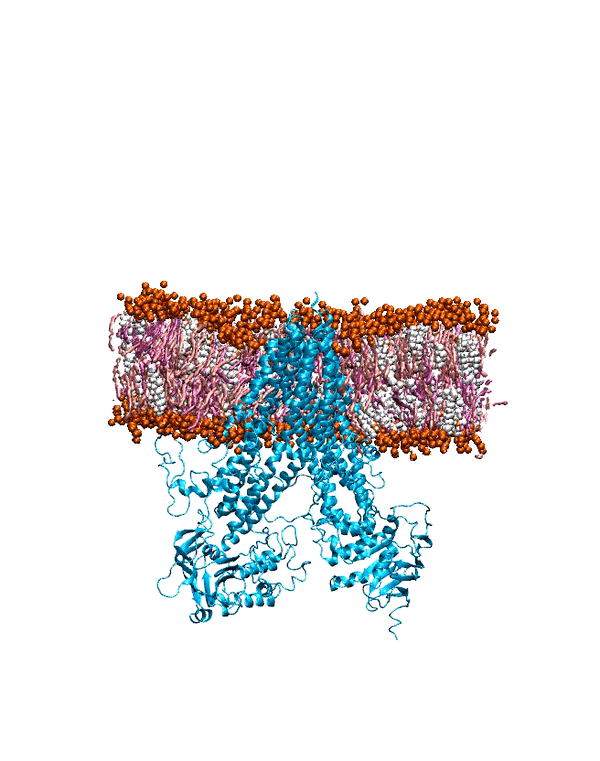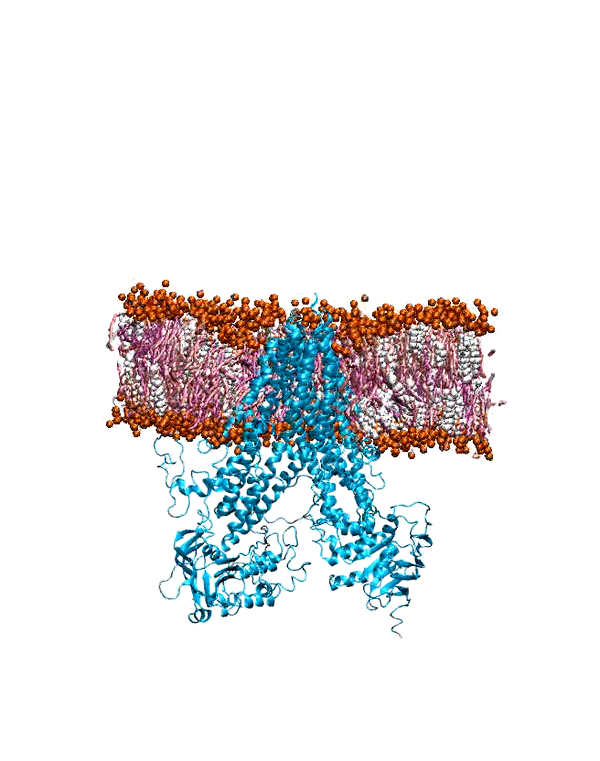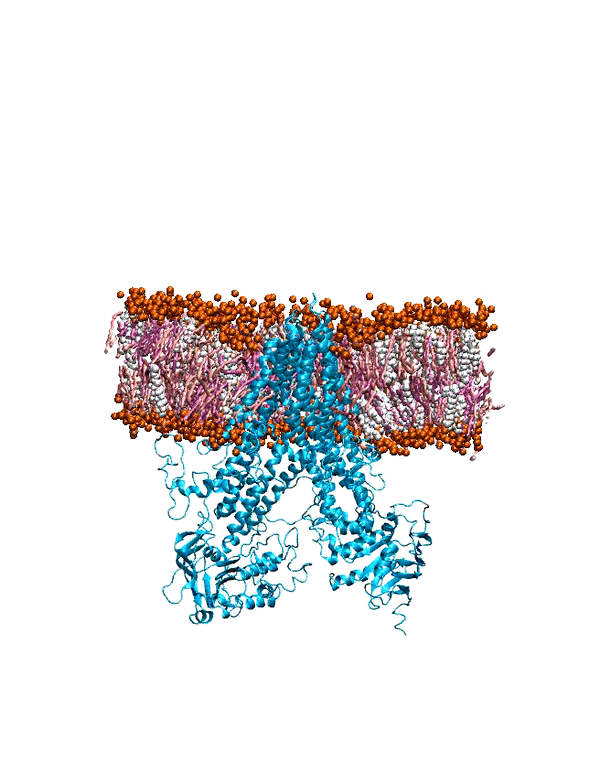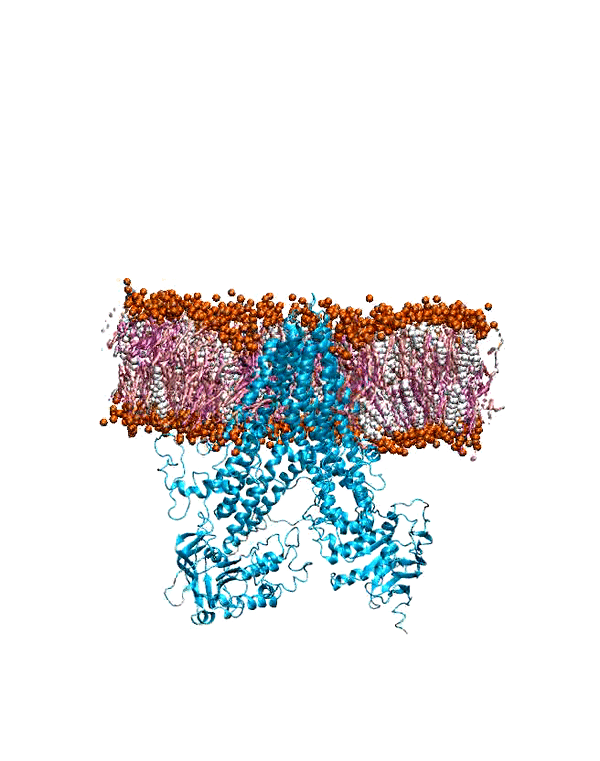
MD Simulation of mGluR1 in Various Cholesterol Concentrations
GPCRs are extremely successful drug targets for treatment of diseases in medicine, with approximately 35% of approved drugs in the pharmaceutical market
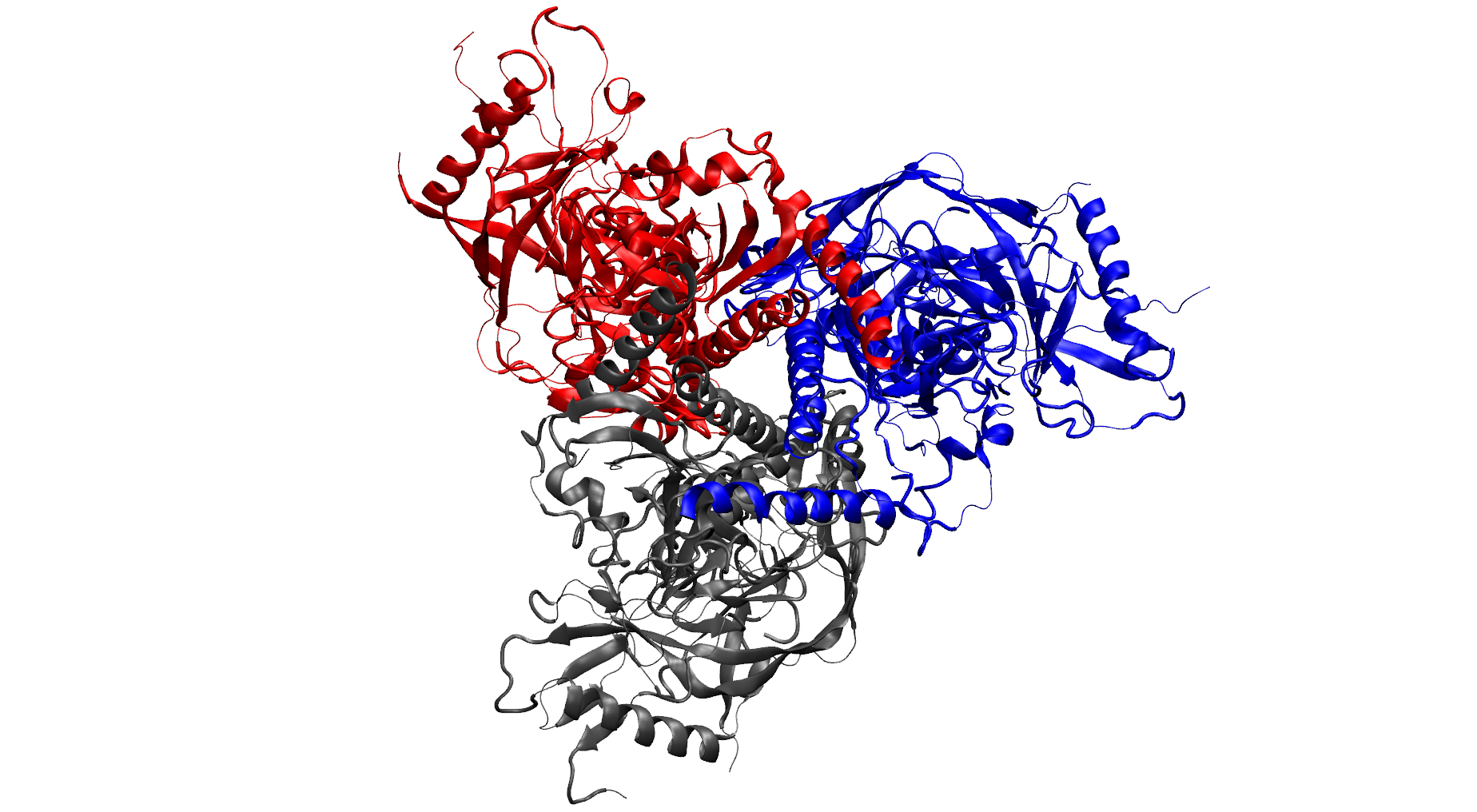
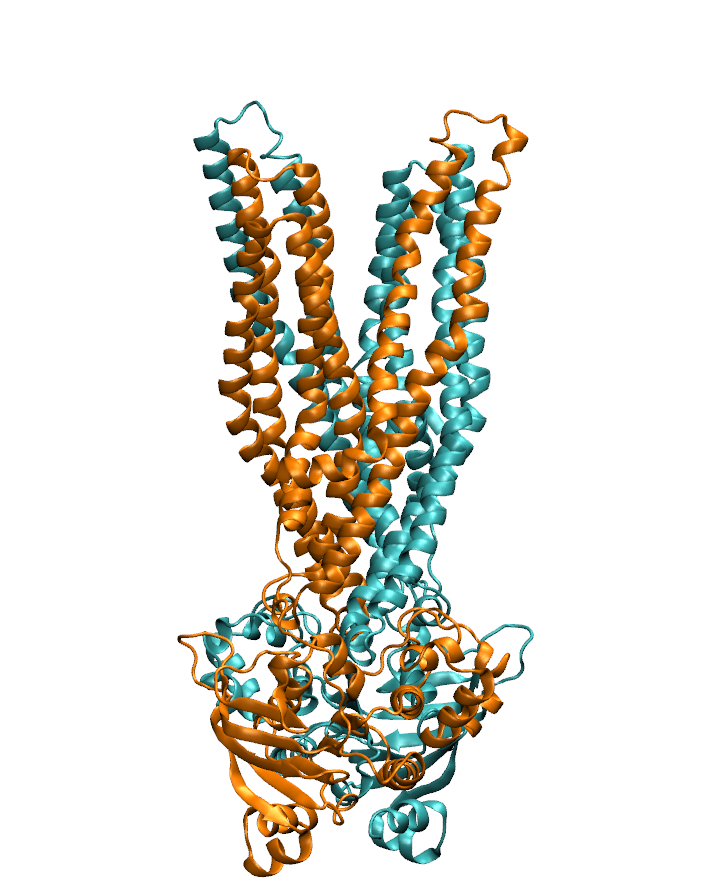
targeting GPCRs. Metabotropic glutamate receptors (mGluRs) are class C G-protein-coupled receptors (GPCRs) which exist as constitutive dimers. They play
significant roles in regulating neurotransmission and activating excitatory synapses in the central nervous system. Besides the common GPCR-defining
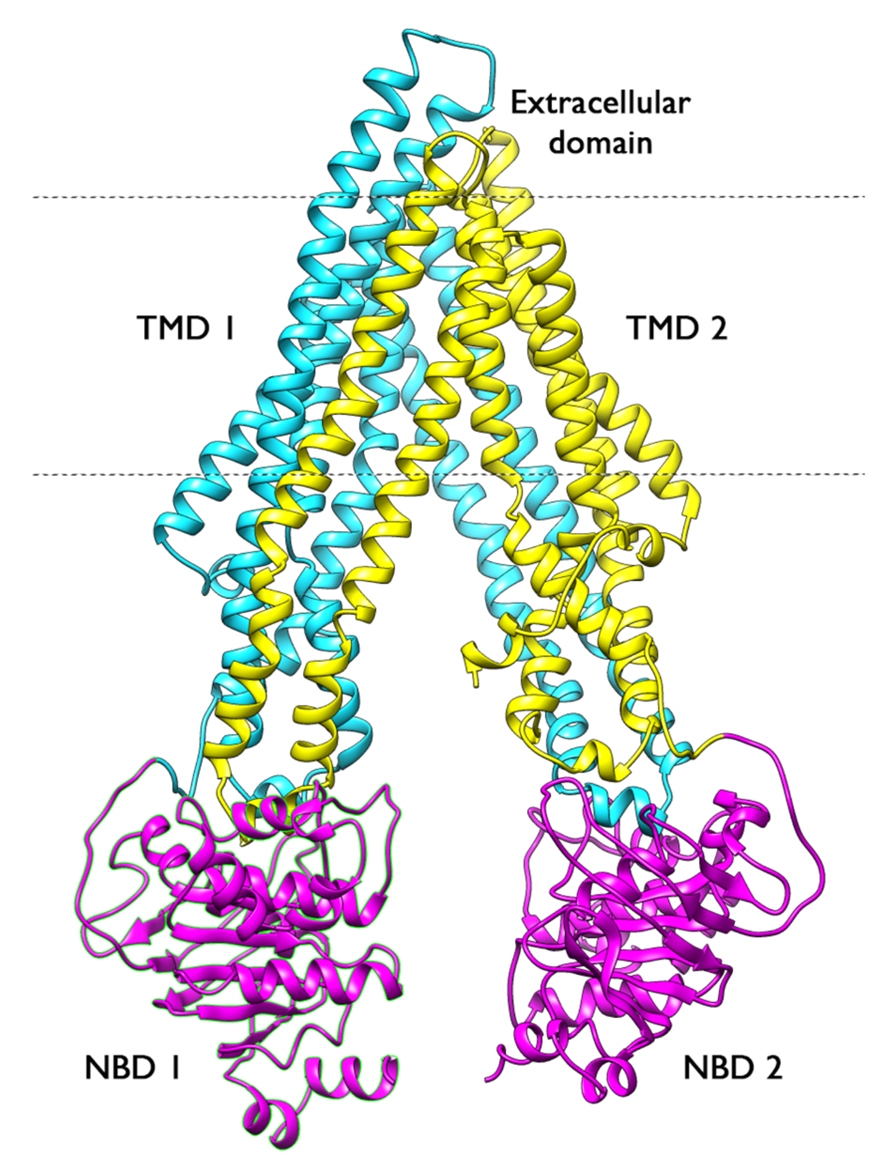
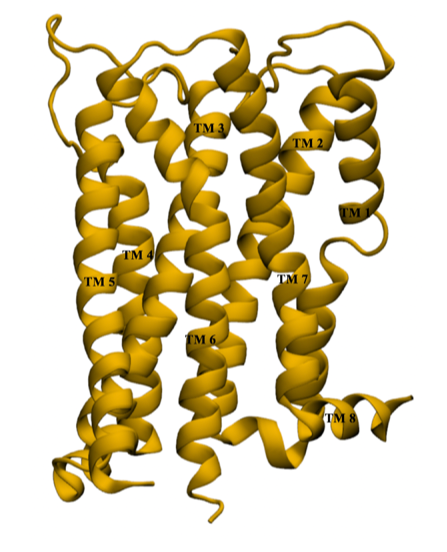
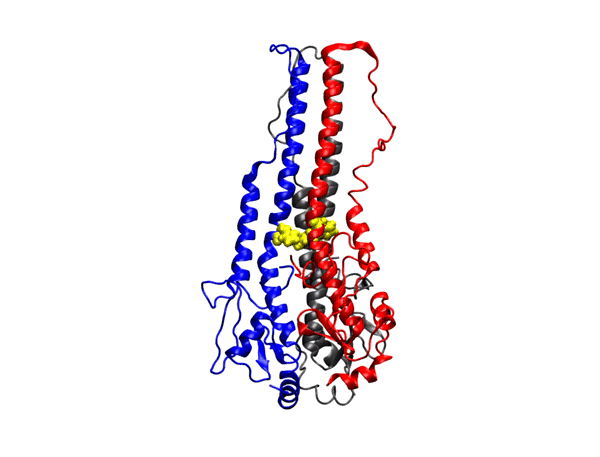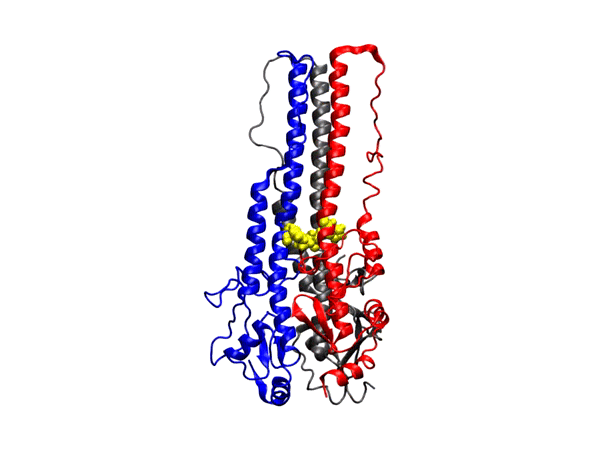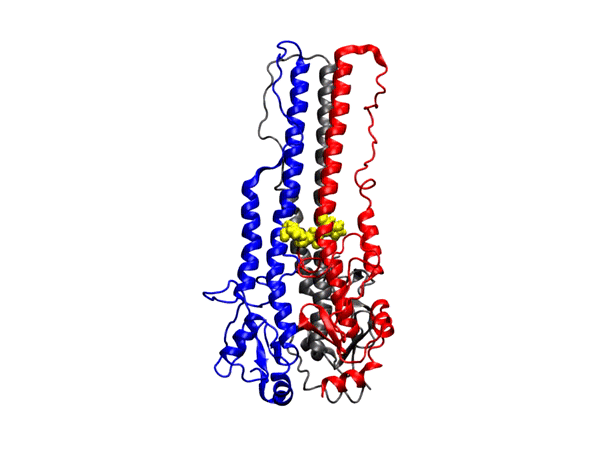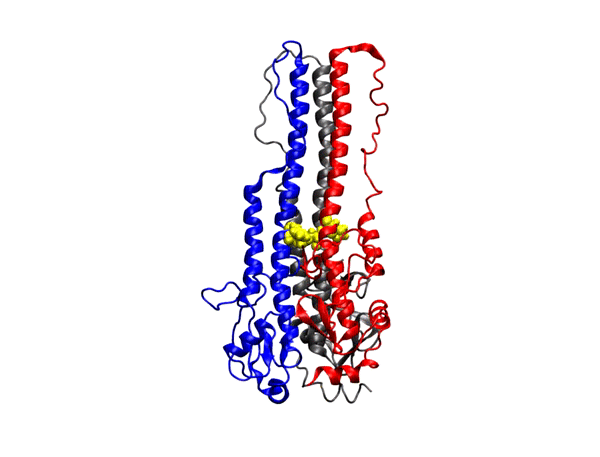
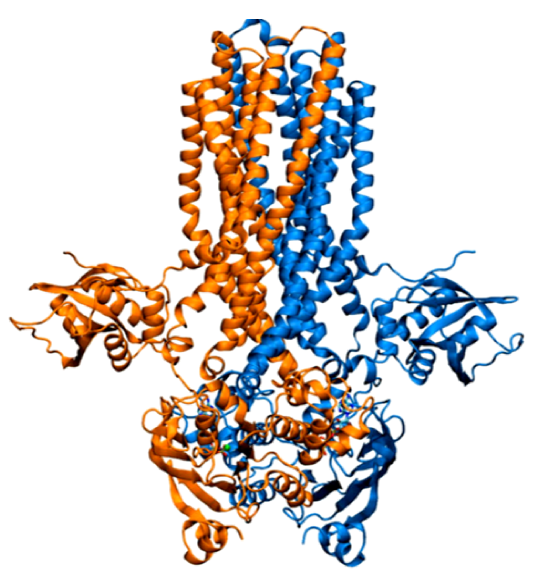
hepta-transmembrane domains (TMDs), the class C GPCRs specifically possess large amino-terminal extracellular domains (ECDs), which comprises of an
orthosteric binding region, a so-called Venus flytrap domain (VFT), and a cysteine-rich domain (CRD). The domains of mGluR1, specifically the 7TM domain
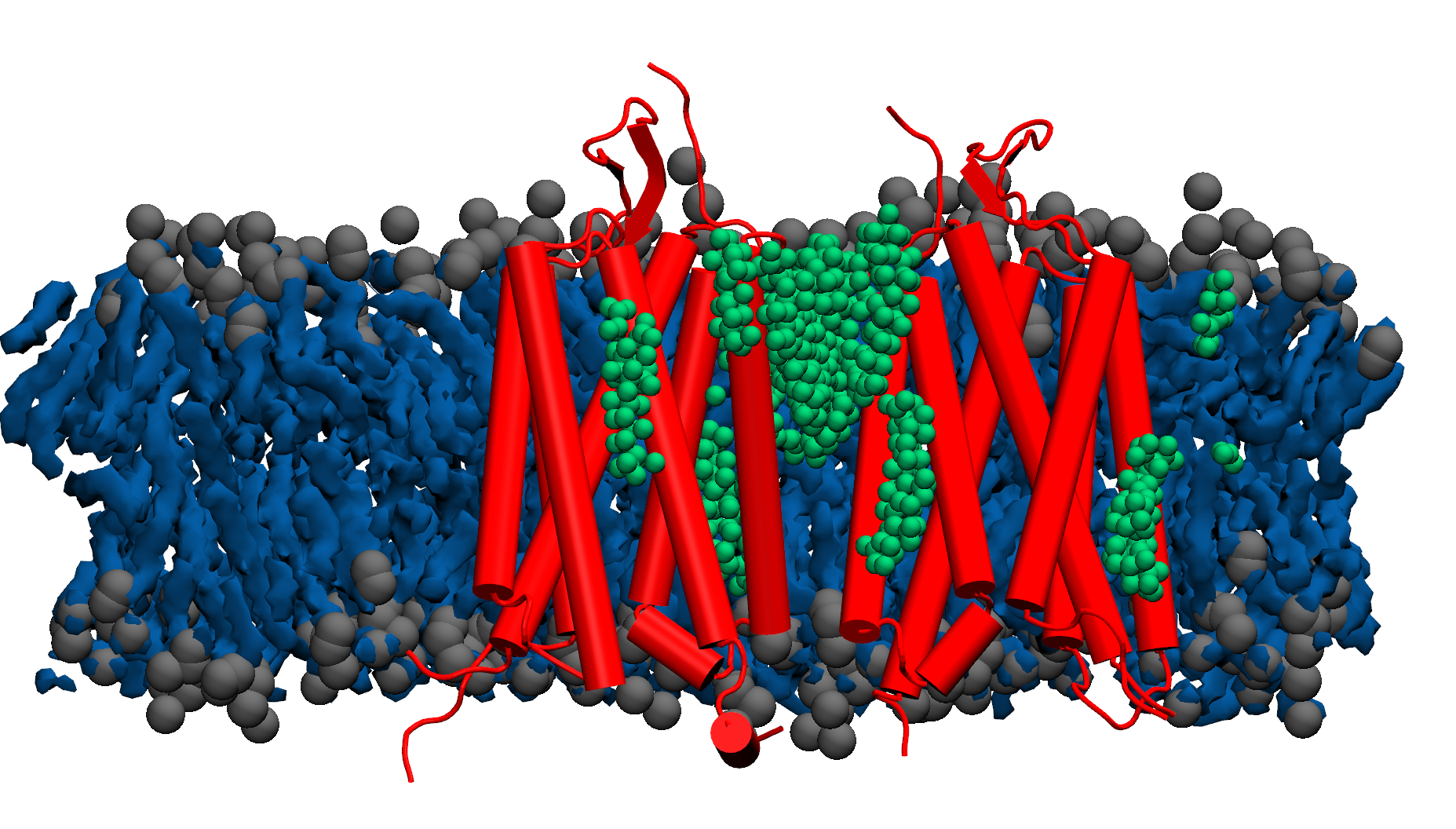
has been shown to be regulated by its surrounding lipid environment, especially by cholesterol although the mechanism of regulation remains elusive.
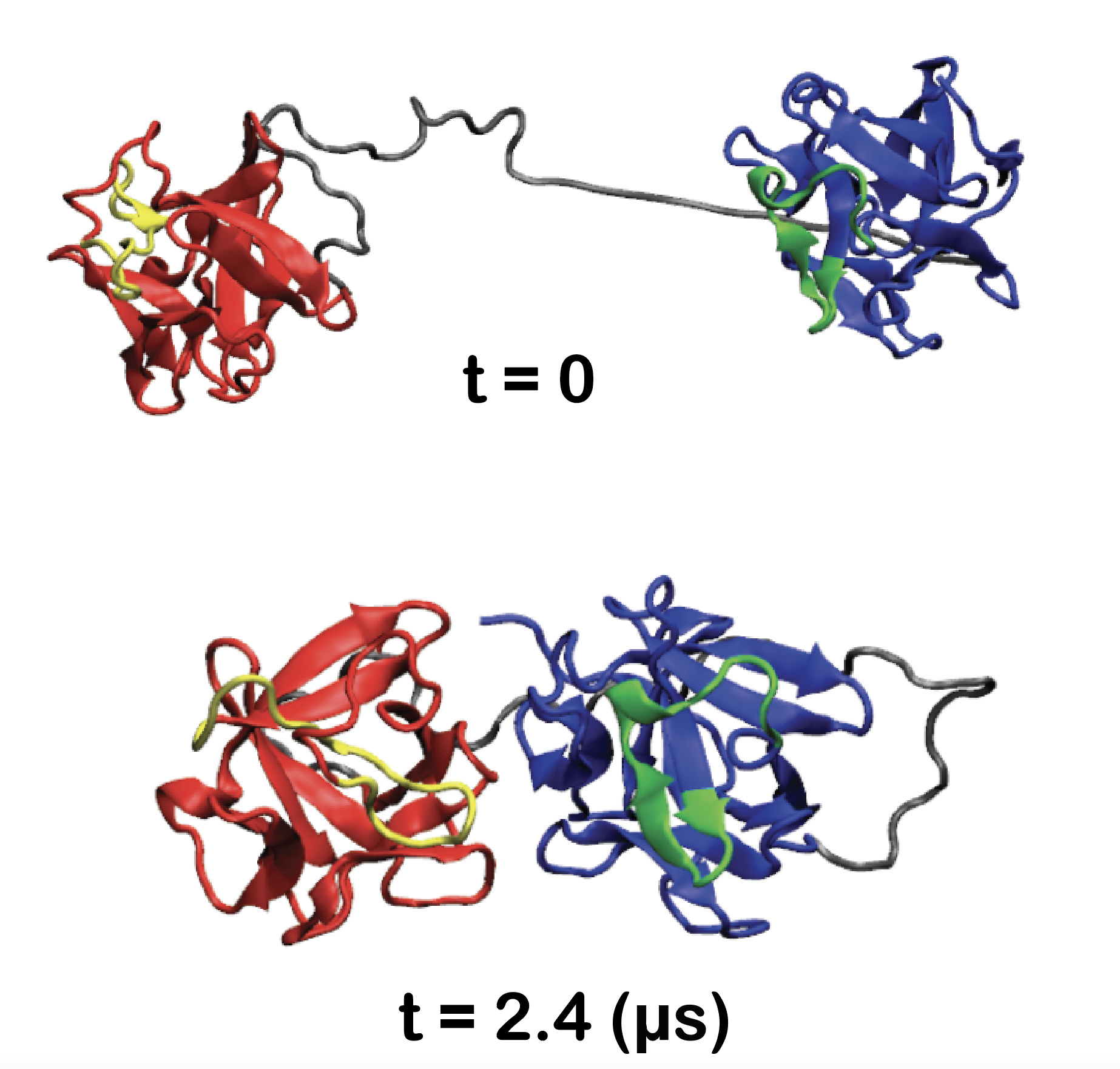
To characterize the conformational dynamics of mGluR1, employing microsecond-level all-atom equilibrium and non-equilibrium MD simulations will enable us
to determine the conformational rearrangements that occur in the different forms of this protein. Understanding and quantifying these functional changes
could lead to the development of potent drugs targeting GPCRs. Due to high sequence conservation of the transmembrane domains (TMDs) of mGluRs, the
molecular interactions observed showing cholesterol dependence in mGluR1 are likely to be applicable for other members of the mGluR family and possibly
GPCR’s in general. Therefore, the goal of this research is to make novel, scientific, and significant contributions to drug development field by
elucidating the activation mechanisms of G-Protein-coupled-receptors (GPCRs).
Our findings have revealed that cholesterol influences the conformational changes of the internal protein and acts less significantly on individual protomers.
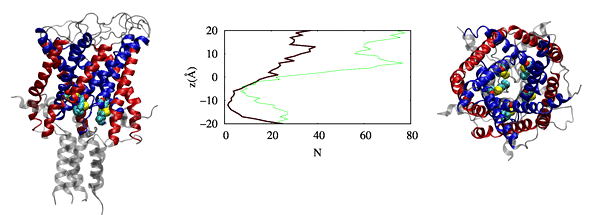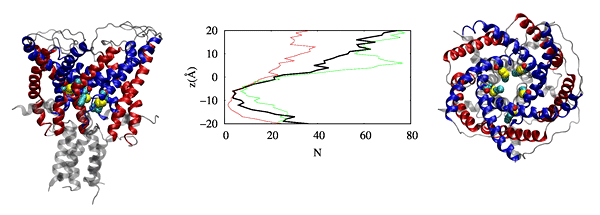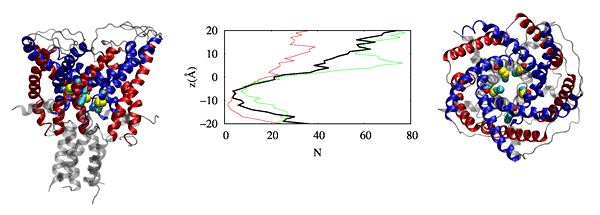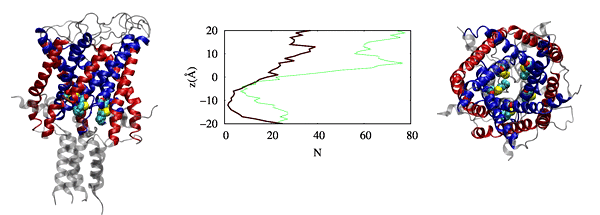
We have also observed that cholesterol affects the dynamics of mGluR1 essentially in TM1 and TM2 which make up the inter-phase of the protein.
Further analysis shows that a low cholesterol concentration induces more significant conformational changes in mGluR1, while the system with higher cholesterol
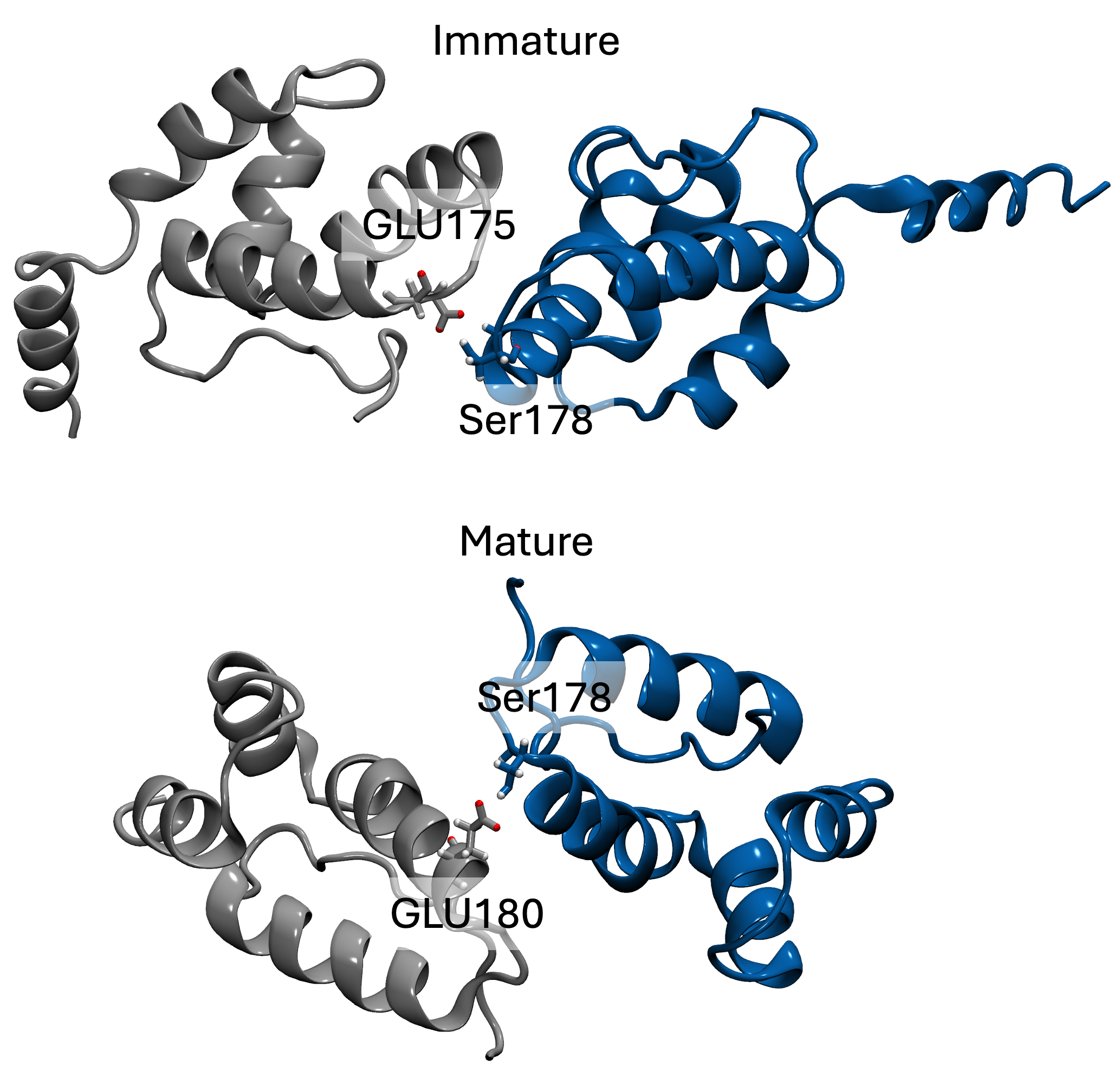
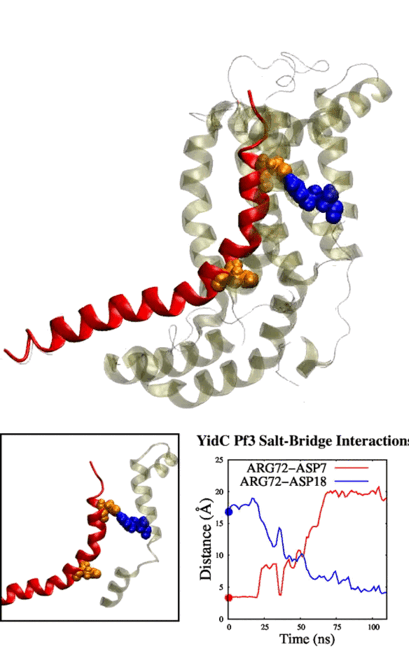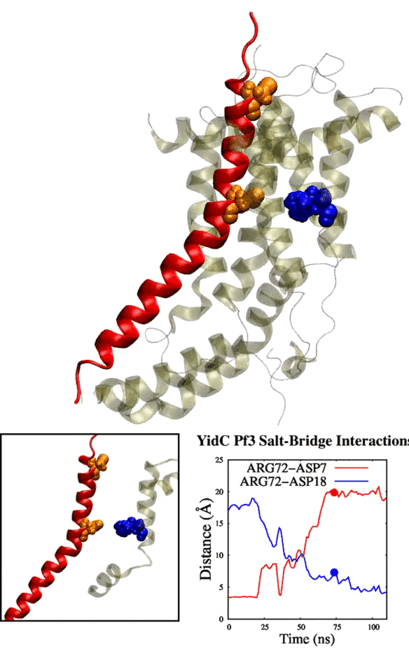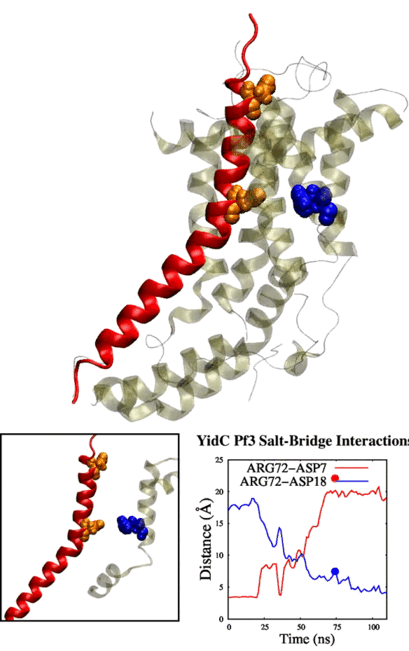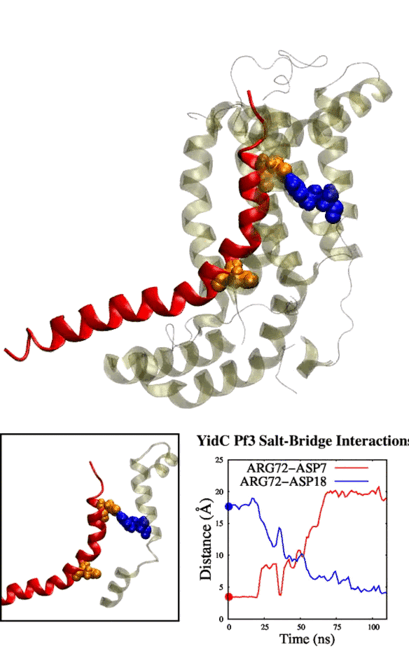
tends to behave similarly to systems without cholesterol. We have also identified some significant electrostatic interactions which are formed only in lower
cholesterol concentration but absent in the other systems.
Conference Poster:
Cholesterol Dependence on the Conformational Changes of Metabotropic Glutamate Receptor 1 (mGluR1)
Conference Poster:
Cholesterol Dependence on the Conformational Changes of Metabotropic Glutamate Receptor 1 (mGluR1)