Research
Quantifying the conformational changes in G–Protein Coupled Receptors using Molecular Dynamics Simulations
G-protein-coupled receptors (GPCRs) are membrane-bound proteins involved in the transmission of external signals to the cell and are involved in vision, taste, olfaction, pain perception, hormone signaling, neurotransmission, etc. Due to their diverse functions, GPCRs are extremely successful drug targets for treatment of common diseases in modern medicine, with approximately 35% of approved drugs in the pharmaceutical market targeting GPCRs.
Experimental methods are able to yield high resolution of these protein structures. However, to fully understand how these proteins interact with each other and their environment, we have to employ computer simulations. Through molecular dynamics simulations, we aim to accurately describe, the structural dynamics of GPCRs in the absence and presence of various membrane lipids (such as cholesterol) and different membrane compositions, and as such develop a computational framework for the rational design of novel therapeutic agents targeting GPCR conformational rearrangements.
Influence of cholesterol on the structural dynamics of metabotropic glutamate receptor (mglur1)
Metabotropic glutamate receptors (mGluRs) are class C, synaptic G-protein-coupled receptors (GPCRs) which consists of a venus fly trap domain, cysteine-rich domain, 7TM domains and form constitutive dimers. The mGluR subfamily has eight members divided into three subgroups (I (mGluR1,5), II (mGluR2,3) and III (mGluR4,6,7,8)) based on their sequence homology, agonist selectivity and signaling function. The group I mGluRs (mGluR1 and mGluR5) are useful for treating diseases such as cancer, pain, schizophrenia, Alzheimer’s disease, anxiety, and autism.
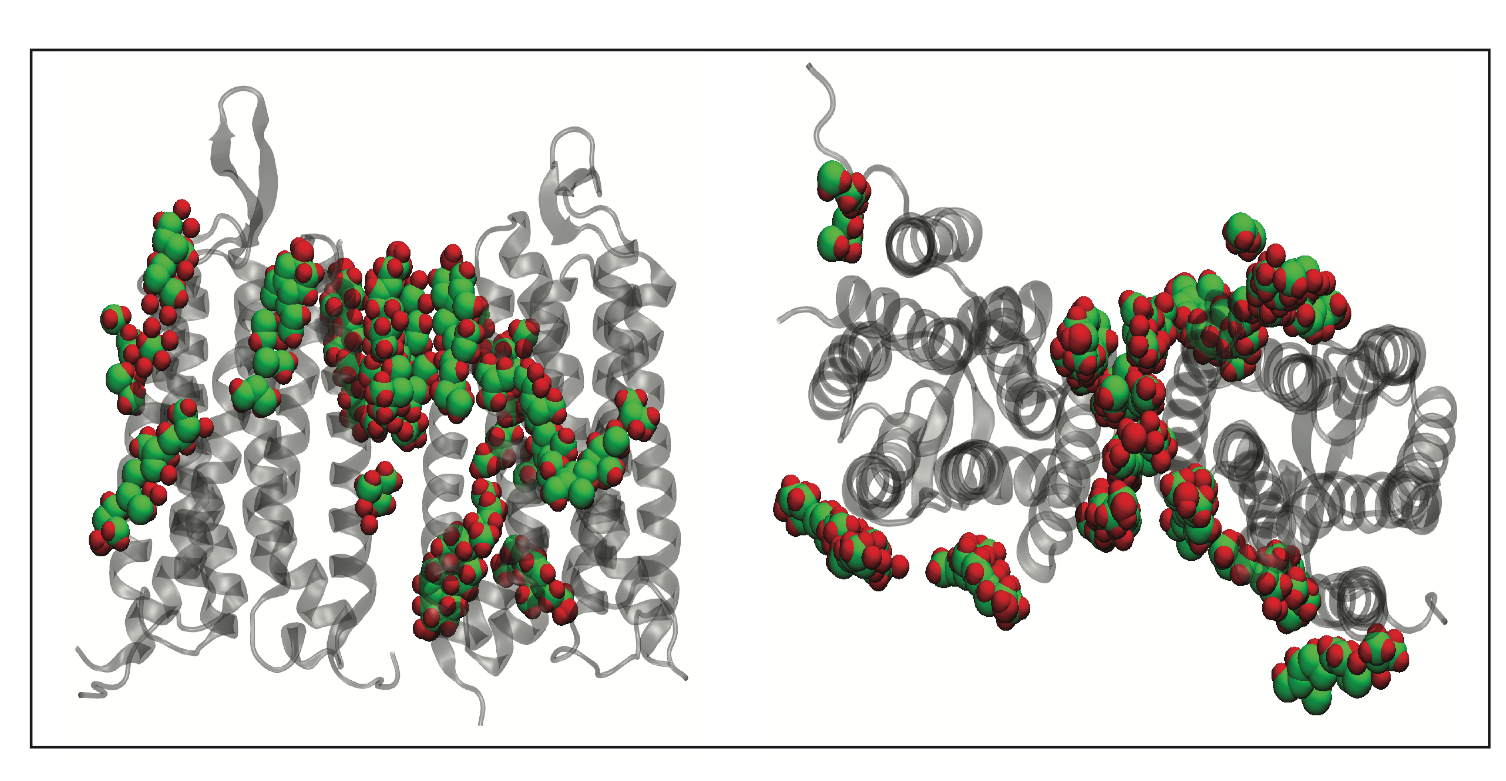
Membrane lipids are major determinants of GPCR organization. Understanding which components of the lipid bilayer are critical in maintaining GPCRs in the appropriate, biologically relevant state is of paramount importance for structure-function studies, and therefore in the design of novel pharmaceuticals. A key multifunctional lipid component of the bilayer is cholesterol. It has been reported experimentally that cholesterol often interacts between dimers of GPCRs to aid dimerization, while in some other GPCRs, the interaction/binding sites are usually between groves of the transmembrane helices. Cholesterol modulates GPCR functions either by direct molecular interaction or indirectly through structural alterations of the physical properties of cell membranes such as membrane fluidity, curvature, lateral pressure and bilayer thickness. Despite several studies ascertaining a likely role for cholesterol in GPCR structure and function, there have been no universal or generalized conclusions, thus
the specific effect of cholesterol on each receptor cannot readily be predicted.
We have used all-atom equilibrium MD simulations to study the effect of membrane cholesterol at varying concentrations, on the local structural dynamics of the mGlur1 7TM region. The preliminary data obtained from 1 microsecond simulation reveals that cholesterol interacts between the dimers of mGluR1 as well as between the groves of the transmembrane helices. We envisage that this study will provide a framework for understanding how cholesterol concentration could drive class C GPCR activation.
|